Introduction
The HistogramZoo R package is a collection of functions which aim to identify and characterize patterns from histogram data.
The Histogram and GenomicHistogram class
A Histogram object contains 1 piece of essential data and 4 pieces of optional data:
histogram_data is a numeric vector representing histogram data. In cases of equal bin width,
histogram_datacan equally represent counts or density with no functional differences. However, in cases of unequal bin width, only density is appropriate for distribution fitting and visualization.interval_start and histogram_end are numeric vectors representing the start and end coordinates of each bin
bin_width is the numeric length of bins. This can be automatically estimated from
interval_startandinterval_end. Objects are required to have equal bin width for every bin with the exception of the last bin, which can have a shorter bin width.region_id is a character id for the histogram
A GenomicHistogram object contains up to 6 additional pieces of data.
chr is a character chromosome
strand is a character strand which is restricted to one of
+,-and*intron_start and intron_end are numeric vectors representing intron coordinates - these can be automatically computed based on non-consecutive
interval_startandinterval_endindicesconsecutive_start and consecutive_end are numeric vectors representing a continuous intronless bin coordinate system on which summary statistics can be computed and distributions can be fitted. Optionally specified by the user,
consecutive_start/endwill be automatically generated based on providedinterval_start/endandintrondata starting at base 1.
A Histogram and a GenomicHistogram differ in several ways:
Conceptualization of bins:
Histogramobjects represent conventional histograms where data are binned on a real number scale. Bins are of the form(start, end]where starts are inclusive while ends are exclusive with the exception of the last bin where both start and end are inclusive.GenomicHistogramobjects were designed to represent a discrete base 1 system where each histogram bin represents a set of indices on which observations are collected.Only
GenomicHistogramobjects support the use of disjoint intervals, or non-consecutive bins. This feature was designed withGenomicHistogramobjects in mind to support the representation of transcripts and genes as a set of exons which might not be consecutive intervals on the genomic coordinate system. Summary statistics and distributions are fit onconsecutive_start/endcoordinates for GenomicHistograms. In operational cases using consecutive bins,consecutive_start/endcan be specified to be the same asinterval_start/end.
my_histogram <- Histogram(
histogram_data = c(1,2,3,1,5,6),
interval_start = c(0,1,2,3,4,5),
interval_end = c(1,2,3,4,5,6),
bin_width = 1,
region_id = "my_histogram"
)
create_coverageplot(
my_histogram,
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
xat = generate_xlabels(my_histogram, n_labels = length(my_histogram), return_xat = T),
xaxis.lab = generate_xlabels(my_histogram, n_labels = length(my_histogram)),
)
my_genomic_histogram <- GenomicHistogram(
histogram_data = c(4,5,2,3,6,8),
interval_start = c(1:3, 5:7),
interval_end = c(1:3, 5:7),
bin_width = 1,
region_id = "my_genomic_histogram",
chr = "chr1",
strand = "*",
intron_start = c(4),
intron_end = c(4),
consecutive_start = seq(6),
consecutive_end = seq(6)
)
create_coverageplot(
my_genomic_histogram,
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
xat = generate_xlabels(my_genomic_histogram, n_labels = length(my_genomic_histogram), return_xat = T),
xaxis.lab = generate_xlabels(my_genomic_histogram, n_labels = length(my_genomic_histogram)),
)
Estimation of summary statistics
Built-in functions can be used to estimate summary statistics, including a weighted mean, variance, standard deviation and skew, on each Histogram object.
histogram_summary <- data.frame(
"id" = "my_histogram",
"mean" = weighted.mean(my_histogram),
"var" = weighted.var(my_histogram),
"sd" = weighted.sd(my_histogram),
"skew" = weighted.skewness(my_histogram)
)
genomic_histogram_summary <- data.frame(
"id" = "my_genomic_histogram",
"mean" = weighted.mean(my_genomic_histogram),
"var" = weighted.var(my_genomic_histogram),
"sd" = weighted.sd(my_genomic_histogram),
"skew" = weighted.skewness(my_genomic_histogram)
)
knitr::kable(
rbind(histogram_summary, genomic_histogram_summary)
)| id | mean | var | sd | skew |
|---|---|---|---|---|
| my_histogram | 3.888889 | 2.722222 | 1.649916 | -0.5943331 |
| my_genomic_histogram | 3.928571 | 3.550265 | 1.884215 | -0.3173915 |
Data loaders
Data loaders for Histogram objects
observations_to_histogram bins a set of observations
at a designated histogram_bin_width to generate a
Histogram
set.seed(314)
my_data <- rnorm(10000, mean = 10, sd = 5)
my_histogram <- observations_to_histogram(my_data, histogram_bin_width = 1)
create_coverageplot(
my_histogram,
main.cex = NULL,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
Data loaders for GenomicHistogram objects
bigWig_to_histogram bins the coverage from a single bigWig file on a set of user-defined regions (provided as a GRanges object, a GRangesList* object or a GTF file) and returns a list of GenomicHistogram objects.
library(GenomicRanges)
filename <- system.file("extdata", "bigwigs", "S1.bw", package = "HistogramZoo")
regions <- GenomicRanges::GRanges(
seqnames = "chr1",
IRanges::IRanges(
start = c(17950, 19350),
end = c(18000, 19600)),
strand = "*"
)
bigwig_histograms <- bigWig_to_histogram(
filename = filename,
regions = regions,
histogram_bin_size = 10
)genome_BED_to_histogram bins the coverage from a set of genomic BED files and returns a list of GenomicHistogram objects. Users have the option of providing a set of regions as a GRanges or GRangesList* object.
datadir <- system.file("extdata", "dna_bedfiles", package = "HistogramZoo")
genome_bed_files <- list.files(datadir, pattern = ".bed$")
genome_bed_files <- file.path(datadir, genome_bed_files)
genome_bed_histograms <- genome_BED_to_histogram(
filenames = genome_bed_files,
n_fields = 6,
histogram_bin_size = 1
)transcript_BED_to_histogram, to account for overlapping transcript annotations, bins the coverage on a set of BED files where each element can be pre-assigned to a region using the 4th column (name) of the BED file.
datadir <- system.file("extdata", "rna_bedfiles", package = "HistogramZoo")
transcript_bed_files <- file.path(datadir, paste0("Sample.", 1:20, ".bed"))
gtf <- system.file("extdata", "genes.gtf", package = "HistogramZoo")
histograms <- transcript_BED_to_histogram(
filenames = transcript_bed_files,
n_fields = 12,
gtf = gtf,
gene_or_transcript = "gene",
histogram_bin_size = 10
)Notes on common parameters
regions
[bigWig, genomic BED]: Using a GRangesList object allows the assignment of disjoint intervals to a GenomicHistogram (e.g. representing a set of exons that can be joined to form a mature transcript) where each GRanges object in the GRangesList represents the elements of a single Histogram. Using a GRanges object creates a separate GenomicHistogram for each element in the GRanges object.histogram_bin_size
[bigWig, genomic and transcriptomic BED]: Bins are generated usingGenomicRanges::tileand coverage is calculated usingGenomicRanges::binnedAverage. This strategy strives to create bins of equal size less than or equal to the designatedhistogram_bin_size(see above for discussion of the difference inbin_widthrestriction.)allow_overlapping_segments_per_sample
[transcriptomic and genomic BED]: Each BED file is assumed to contain the elements of a single experimental sample. Thus, overlapping elements in each BED file can be eliminated by setting the parameter toFALSE.
Segmentation and distribution fitting
The function segment_and_fit is designed to be a
pipeline constructed from the following sub-functions which enables the
segmentation, denoising and fitting of statistical distributions on
Histogram and GenomicHistogram objects. Each of the parameters in
segment_and_fit have counterparts in the sub-functions
which is explored in further detail below.
NOTA BENE: Most of the functions below are
implemented as S3 methods which can take Histogram,
GenomicHistogram and numeric vector objects as
input.
Finding local optima
find_local_optima identifies the local minima and maxima on a vector of counts (histogram data).
# Generating a Histogram
set.seed(314)
dt = rnorm(10000, mean = 20, sd = 10)
xhist = observations_to_histogram(dt, histogram_bin_width = 2)
# basic use
optima = find_local_optima(xhist)
optima = sort(unlist(optima))
create_coverageplot(
xhist,
add.points = T,
points.x = optima,
points.y = xhist$histogram_data[optima],
points.col = "red",
main = "basic optima",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
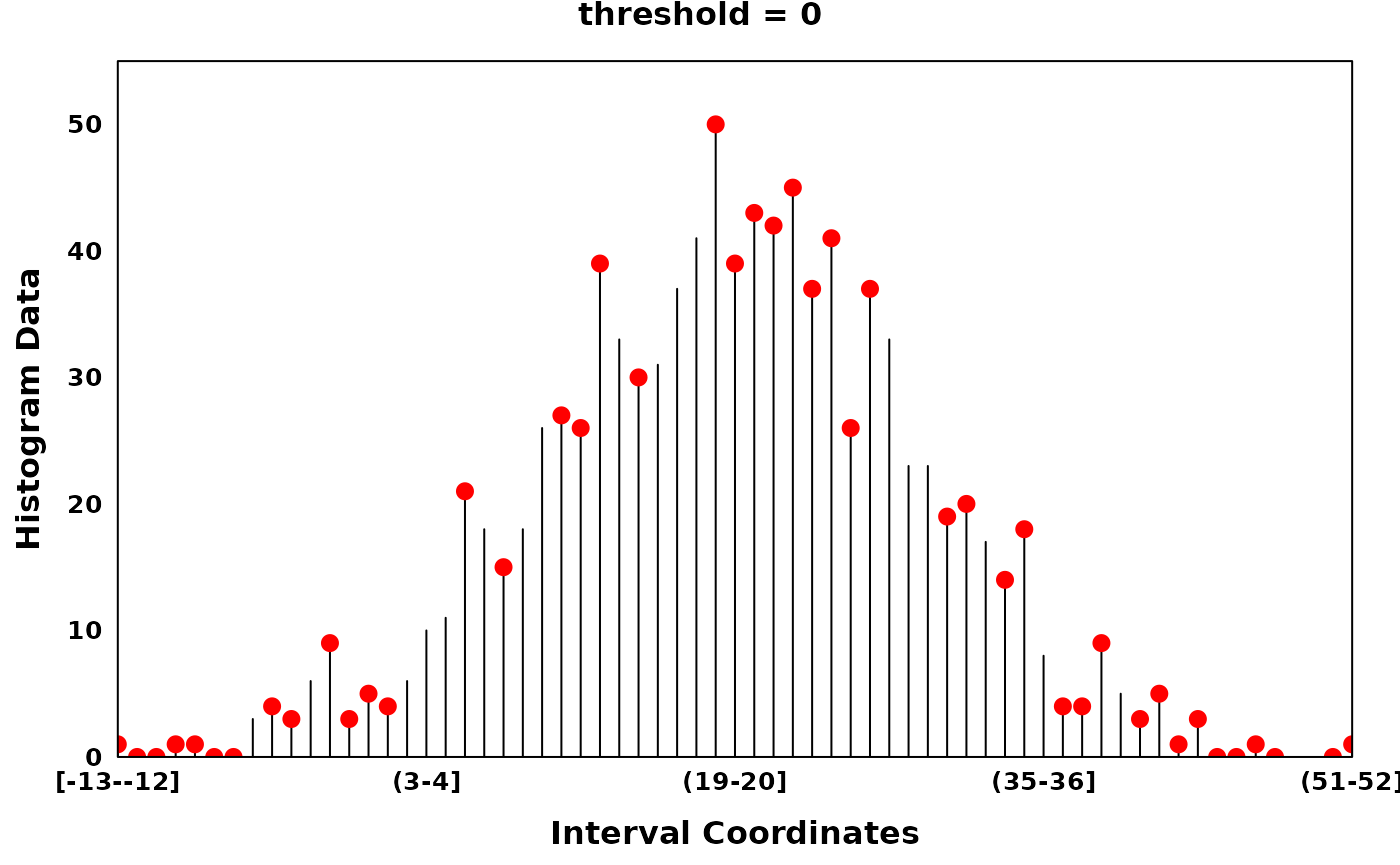
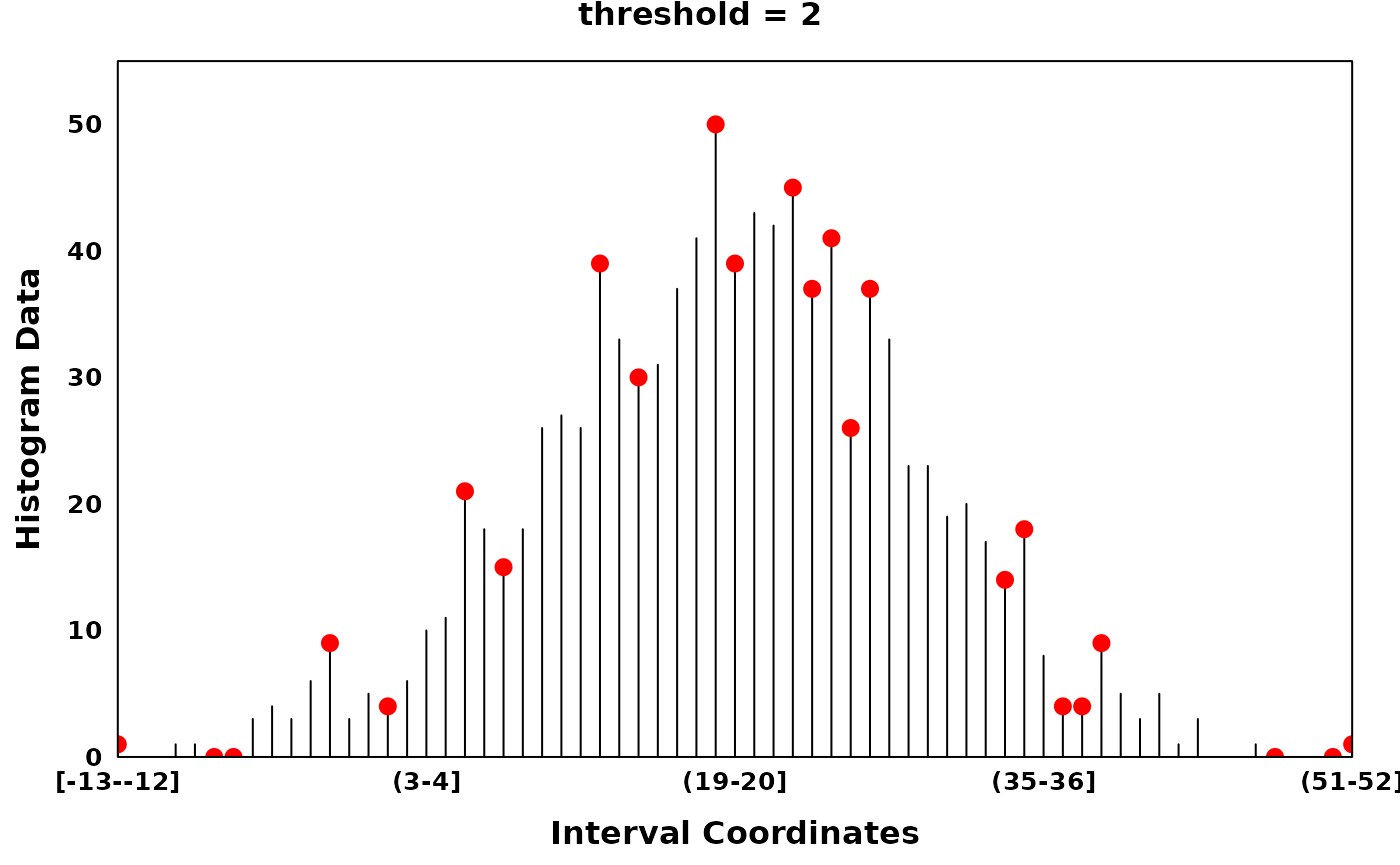
The parameter threshold allows the user to tune the
sparsity of local optima to histograms with potentially noisy counts,
i.e. by thresholding the difference between neighbouring optima.
# Generating a Histogram with noisy counts
set.seed(314)
dt = rnorm(1000, mean = 20, sd = 10)
xhist = observations_to_histogram(dt, histogram_bin_width = 1)
# find_local_optima with threshold 0
zero_threshold_optima = find_local_optima(xhist, threshold = 0)
zero_threshold_optima = sort(unlist(zero_threshold_optima))
create_coverageplot(
xhist,
add.points = T,
points.x = zero_threshold_optima,
points.y = xhist$histogram_data[zero_threshold_optima],
points.col = "red",
type = "h",
main = "threshold = 0",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
# find_local_optima with threshold 2
threshold_optima = find_local_optima(xhist, threshold = 2)
threshold_optima = sort(unlist(threshold_optima))
create_coverageplot(
xhist,
add.points = T,
points.x = threshold_optima,
points.y = xhist$histogram_data[threshold_optima],
points.col = "red",
type = "h",
main = "threshold = 2",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
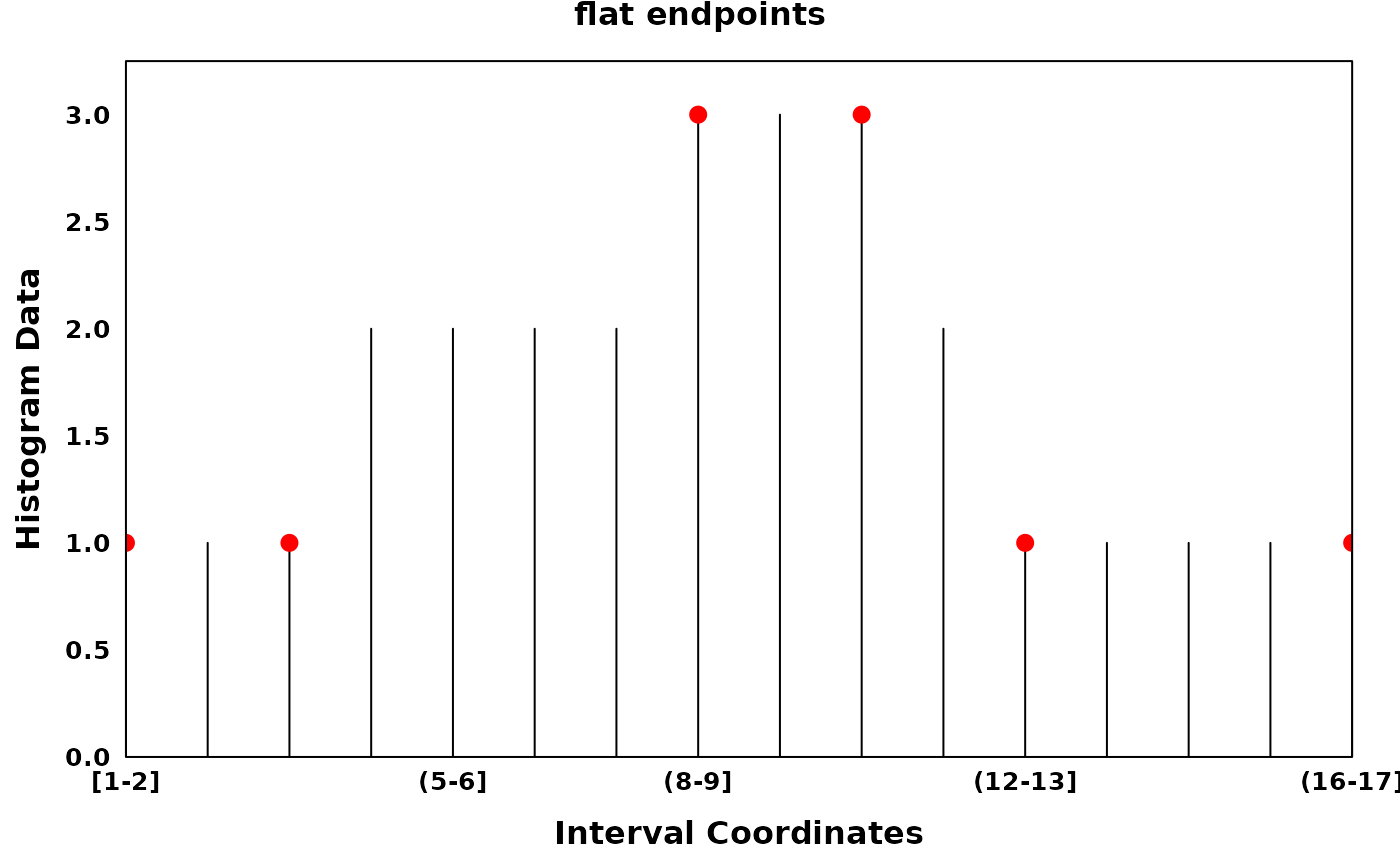
The parameter flat_endpoints allows the user to better
fit histograms with stretches of equal values (e.g. pile-ups of BED
files in a GenomicHistogram). Setting the parameter to TRUE
will allow the user to return both endpoints of a flat region while
setting the parameter to FALSE will return the estimated
midpoint of a flat region.
# Generating a Histogram with consecutive equal values
data = c(rep(1, 3), rep(2, 4), rep(3, 3), rep(2, 1), rep(1, 5))
xhist = Histogram(histogram_data = data)
# flat_endpoints
optima_flat = find_local_optima(xhist, flat_endpoints = TRUE)
optima_flat = sort(unlist(optima_flat))
create_coverageplot(
xhist,
add.points = T,
points.x = optima_flat,
points.y = xhist$histogram_data[optima_flat],
points.col = "red",
main = "flat endpoints",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
# midpoints
optima_midpoints = find_local_optima(xhist, flat_endpoints = FALSE)
optima_midpoints = sort(unlist(optima_midpoints))
create_coverageplot(
xhist,
add.points = T,
points.x = optima_midpoints,
points.y = xhist$histogram_data[optima_midpoints],
points.col = "red",
main = "midpoints",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)

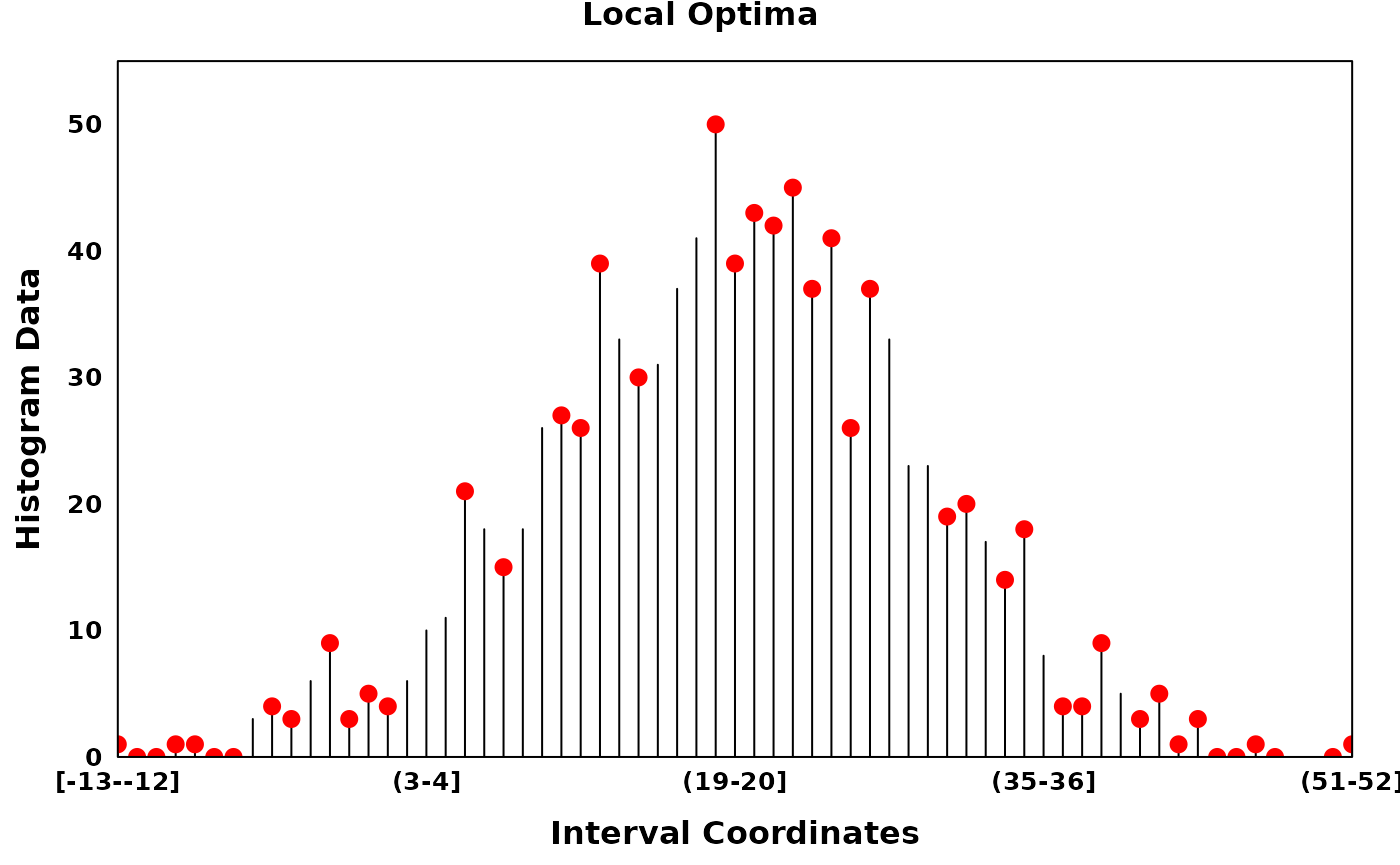
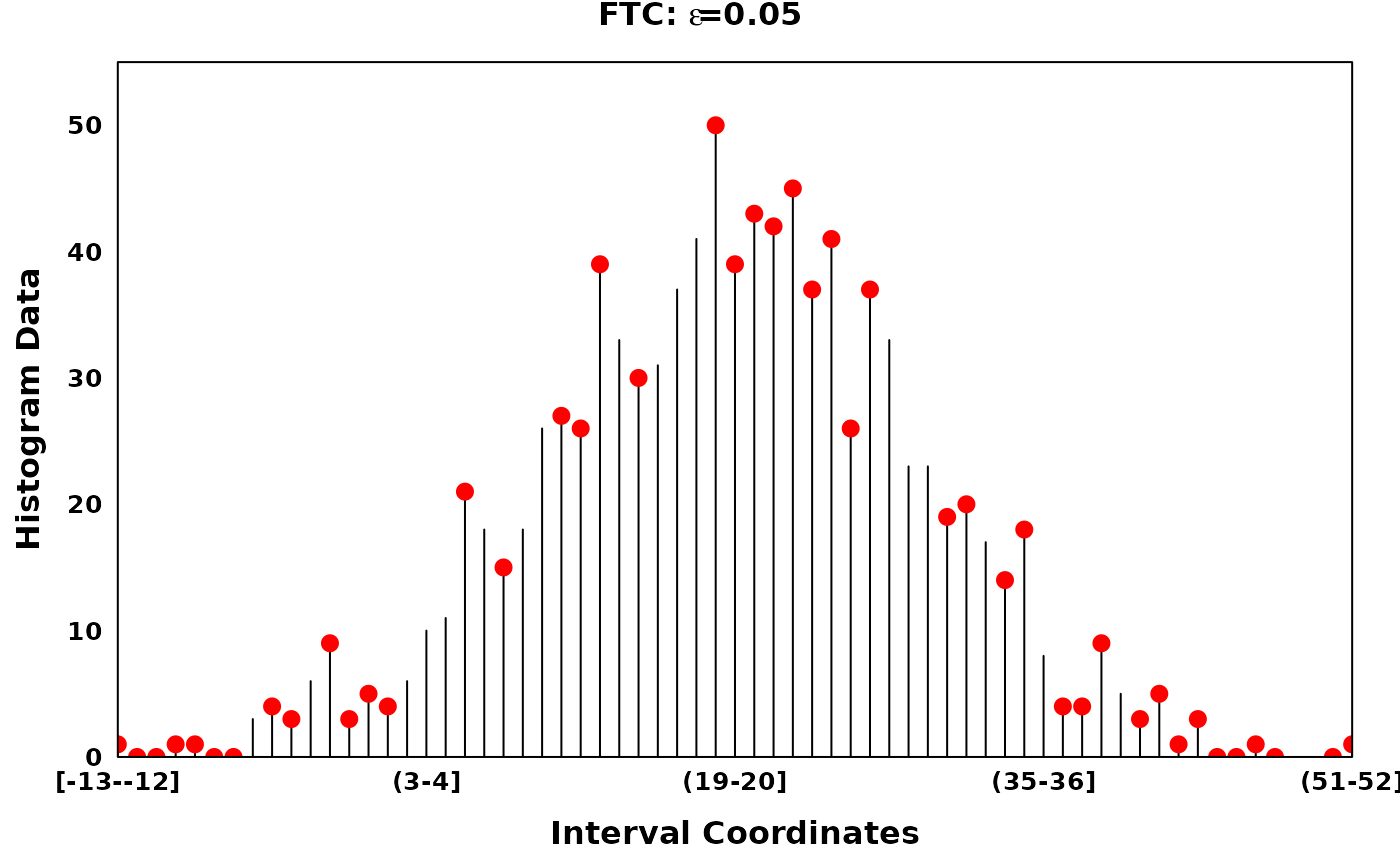
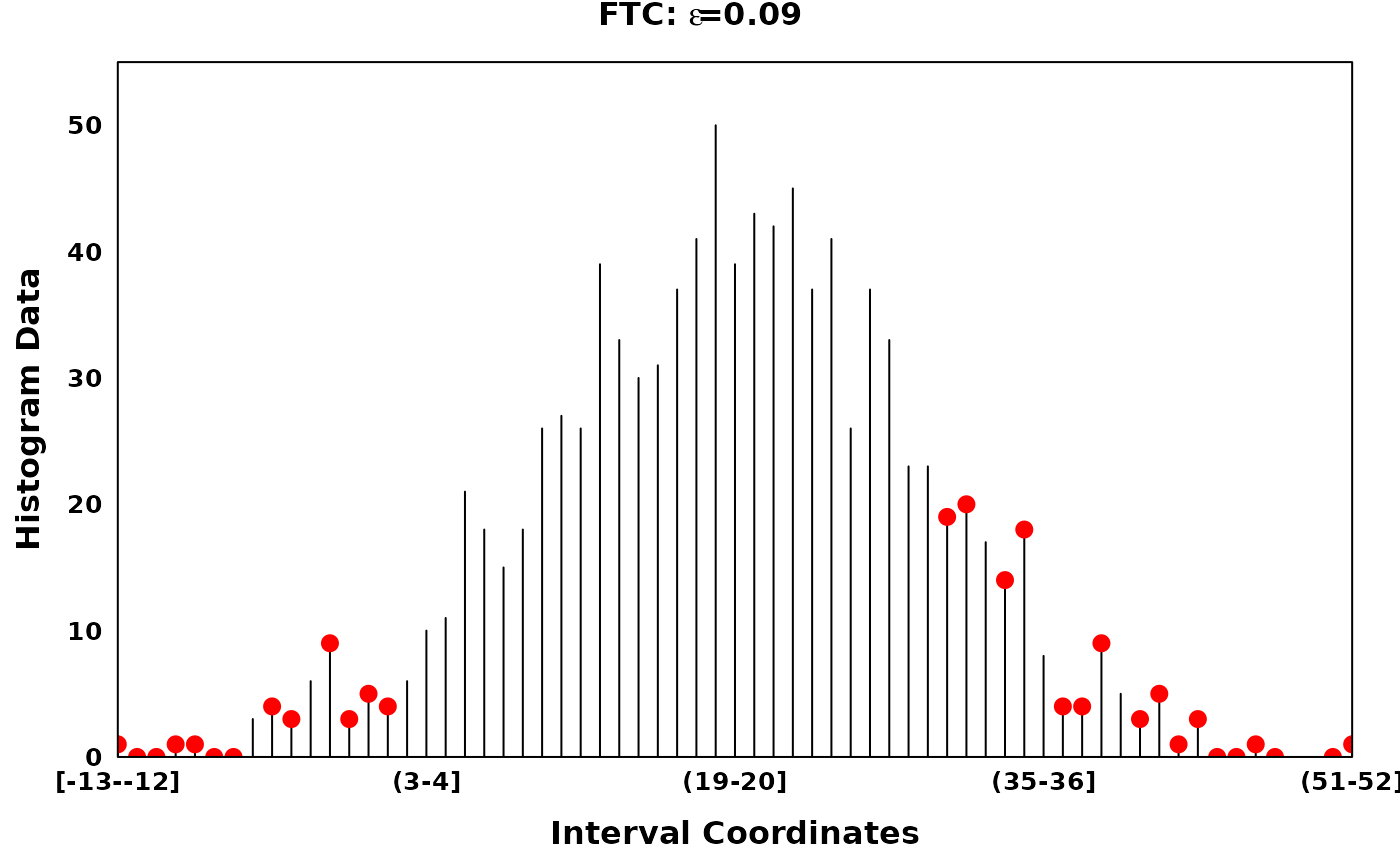
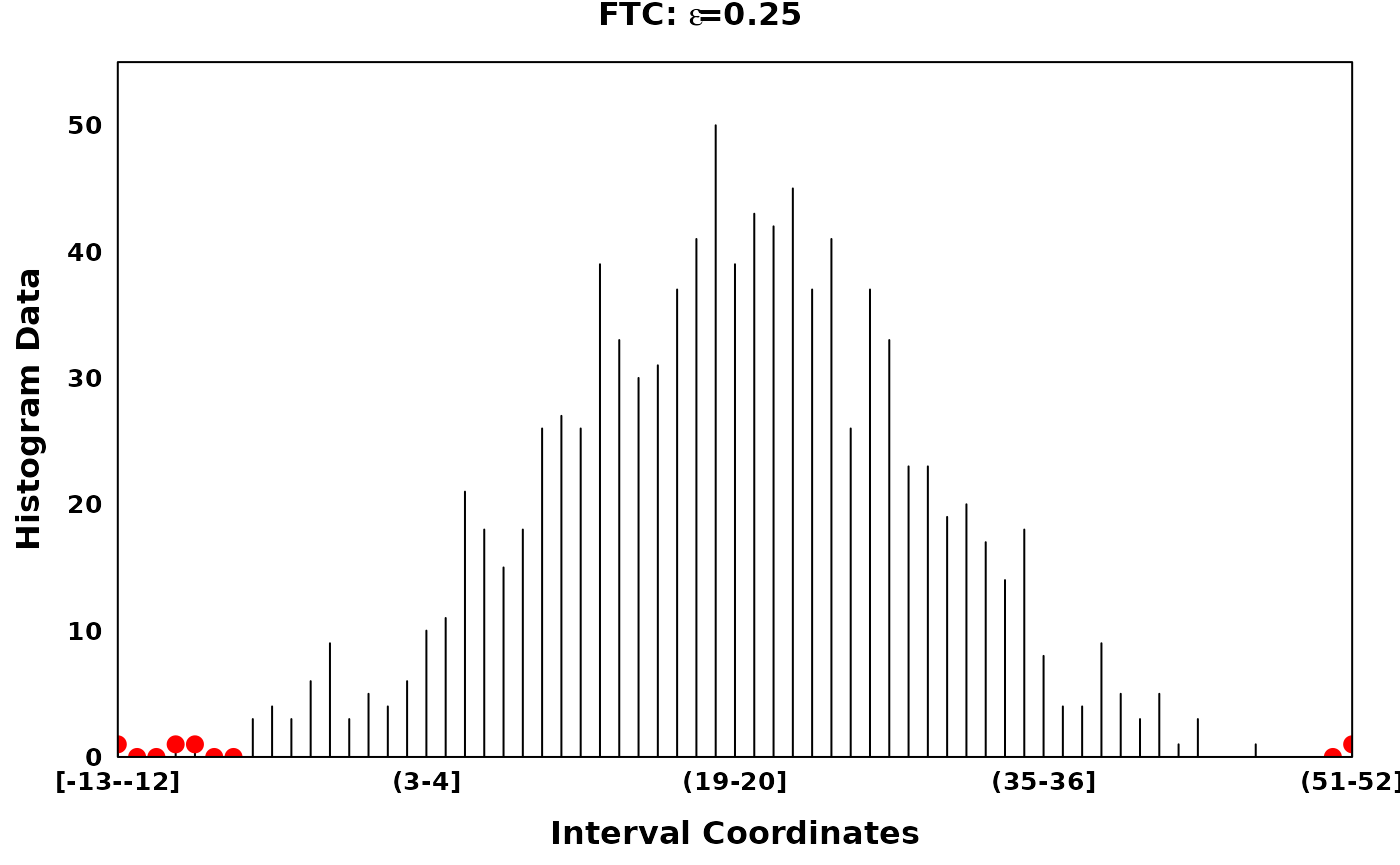
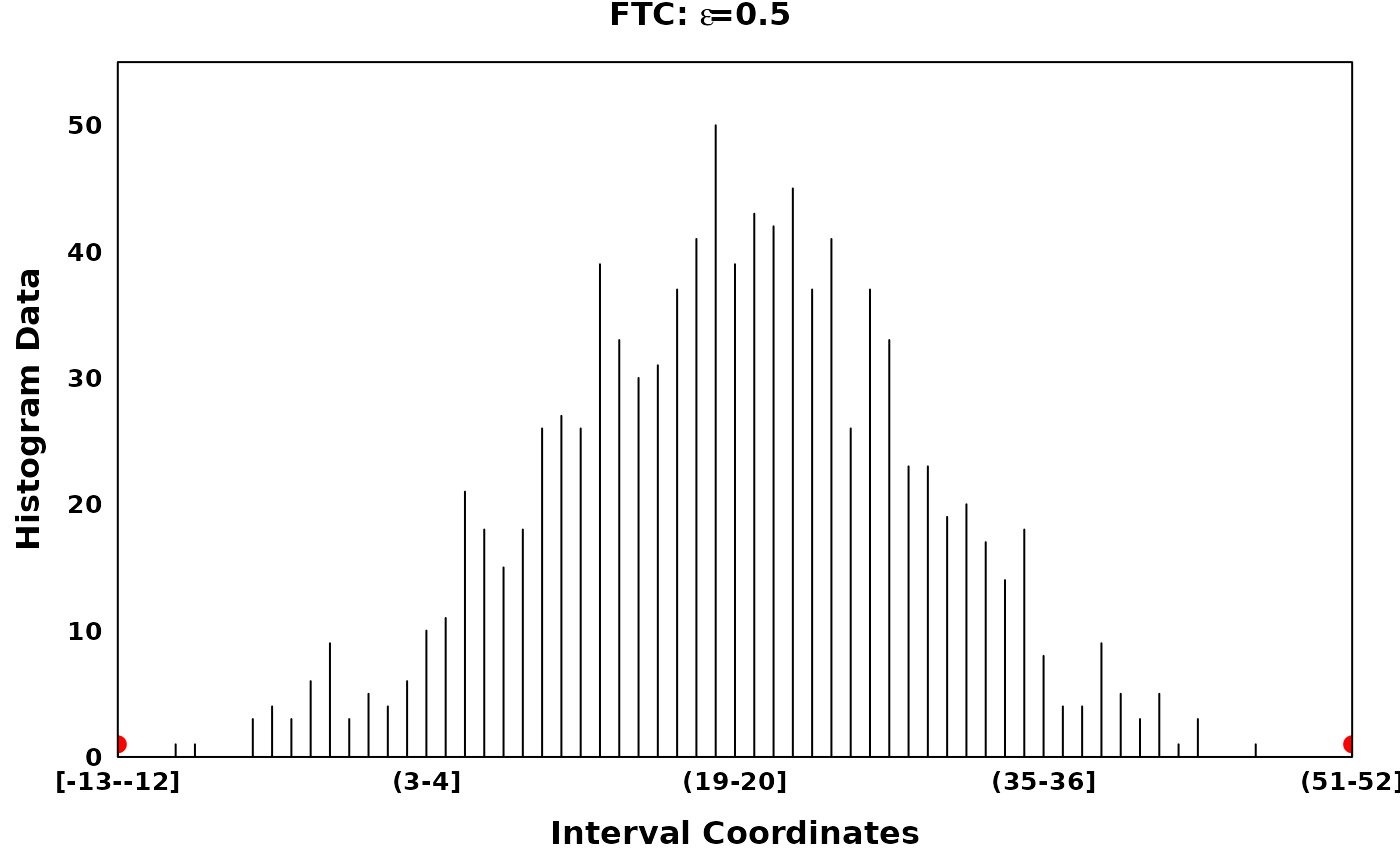
The Fine-to-Coarse segmentation algorithm
The Fine-to-Coarse segmentation algorithm was described in Delon et al. (2005), Lisani and Petro (2021) and Delon et al. (2006). Briefly, for a given histogram and a set of local optima, the FTC algorithm iterates through consecutive segments identifying peaks, segments of increasing counts followed by segments of decreasing counts. Consecutive segments are merged based on the prioritization derived from a monotone cost function, until an optimal set of peaks are acquired. Granularity of the optimal peak set can be tuned via the hyperparamter \(\epsilon\).
# Generating a Histogram with noisy counts
set.seed(314)
dt = rnorm(1000, mean = 20, sd = 10)
xhist = observations_to_histogram(dt, histogram_bin_width = 1)
# local optima
zero_threshold_optima = find_local_optima(xhist, threshold = 0)
zero_threshold_optima = sort(unlist(zero_threshold_optima))
create_coverageplot(
xhist,
add.points = T,
points.x = zero_threshold_optima,
points.y = xhist$histogram_data[zero_threshold_optima],
points.col = "red",
type = "h",
main = "Local Optima",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
# Using different epsilons on FTC
ftc_eps_0.05 <- ftc(xhist, s = zero_threshold_optima, eps = 0.05)
ftc_eps_0.09 <- ftc(xhist, s = zero_threshold_optima, eps = 0.09)
ftc_eps_0.15 <- ftc(xhist, s = zero_threshold_optima, eps = 0.15)
ftc_eps_0.25 <- ftc(xhist, s = zero_threshold_optima, eps = 0.25)
ftc_eps_0.5 <- ftc(xhist, s = zero_threshold_optima, eps = 0.5)
create_coverageplot(
xhist,
add.points = T,
points.x = ftc_eps_0.05,
points.y = xhist$histogram_data[ftc_eps_0.05],
points.col = "red",
type = "h",
main = expression(bold(paste("FTC: ", epsilon, "=0.05"))),
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
create_coverageplot(
xhist,
add.points = T,
points.x = ftc_eps_0.09,
points.y = xhist$histogram_data[ftc_eps_0.09],
points.col = "red",
type = "h",
main = expression(bold(paste("FTC: ", epsilon, "=0.09"))),
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
create_coverageplot(
xhist,
add.points = T,
points.x = ftc_eps_0.15,
points.y = xhist$histogram_data[ftc_eps_0.15],
points.col = "red",
type = "h",
main = expression(bold(paste("FTC: ", epsilon, "=0.15"))),
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
create_coverageplot(
xhist,
add.points = T,
points.x = ftc_eps_0.25,
points.y = xhist$histogram_data[ftc_eps_0.25],
points.col = "red",
type = "h",
main = expression(bold(paste("FTC: ", epsilon, "=0.25"))),
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
create_coverageplot(
xhist,
add.points = T,
points.x = ftc_eps_0.5,
points.y = xhist$histogram_data[ftc_eps_0.5],
points.col = "red",
type = "h",
main = expression(bold(paste("FTC: ", epsilon, "=0.5"))),
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)

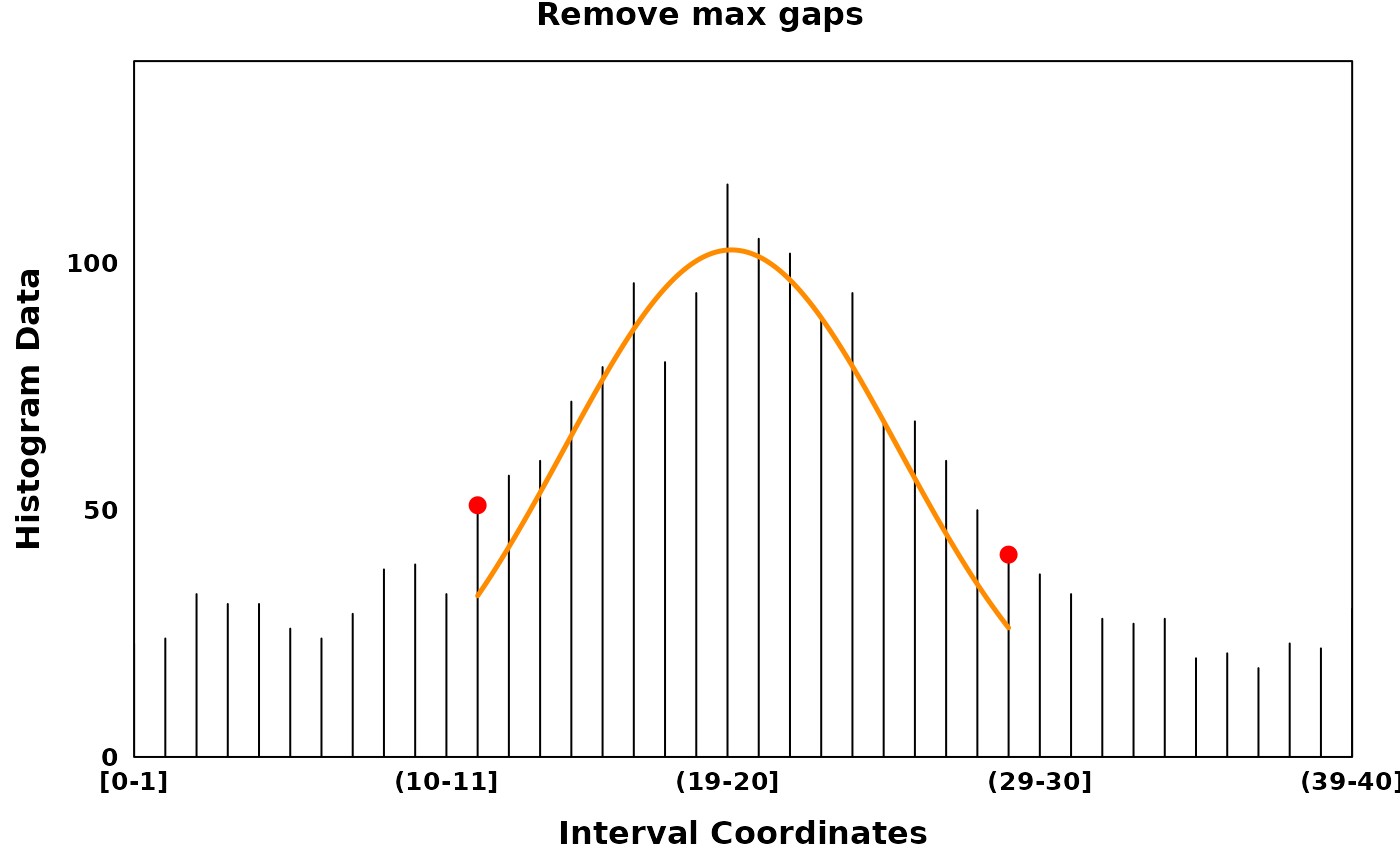
Filtering low entropy regions
In the presence of noisy data, it can be helpful to filter regions
with low entropy to extract high density regions. Low entropy regions or
max_gaps can be identified using the algorithm described in
Delon et al. (2005). Briefly, a region is
defined as a gap if it has high entropy and low density relative to its
adjacent peak. Identification of max gaps is conducted on each segment
post-segmentation.
# Generating a Histogram with noisy counts
set.seed(314)
dt = c(
rnorm(1000, mean = 20, sd = 5),
runif(1000, min = 0, max = 40)
)
xhist = observations_to_histogram(dt, histogram_bin_width = 1)
# Segment and Fit with and without filtering for max gaps
filtered_fit <- segment_and_fit(
histogram_obj = xhist,
eps = 1,
seed = 314,
remove_low_entropy = TRUE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
unfiltered_fit <- segment_and_fit(
histogram_obj = xhist,
eps = 1,
seed = 314,
remove_low_entropy = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
unfiltered_plt <- create_coverageplot(
unfiltered_fit,
main = "Unfiltered",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
# Plotting
filtered_plt <- create_coverageplot(
filtered_fit,
main = "Remove max gaps",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(unfiltered_plt)
print(filtered_plt)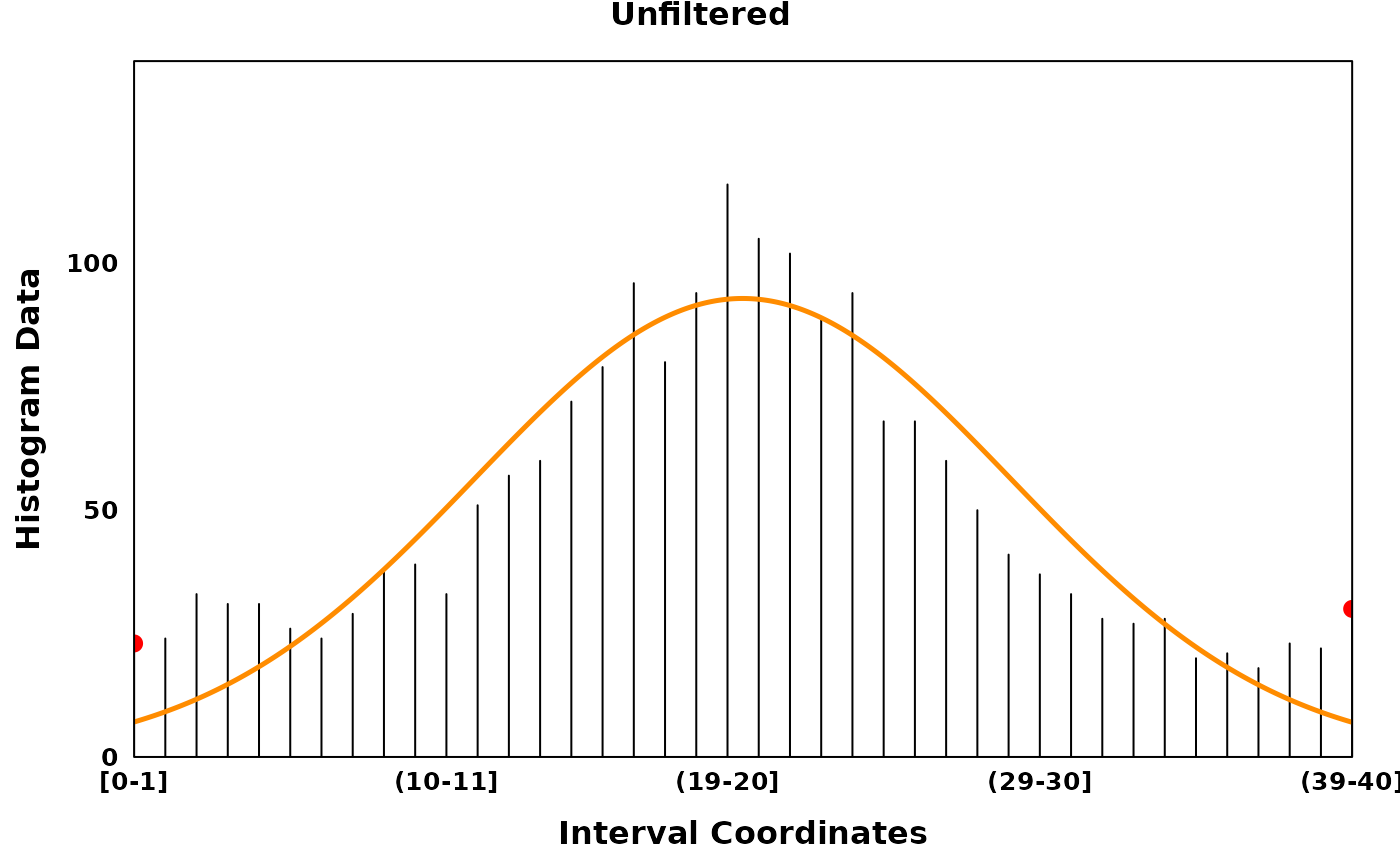
Fitting statistical distributions
Distributions
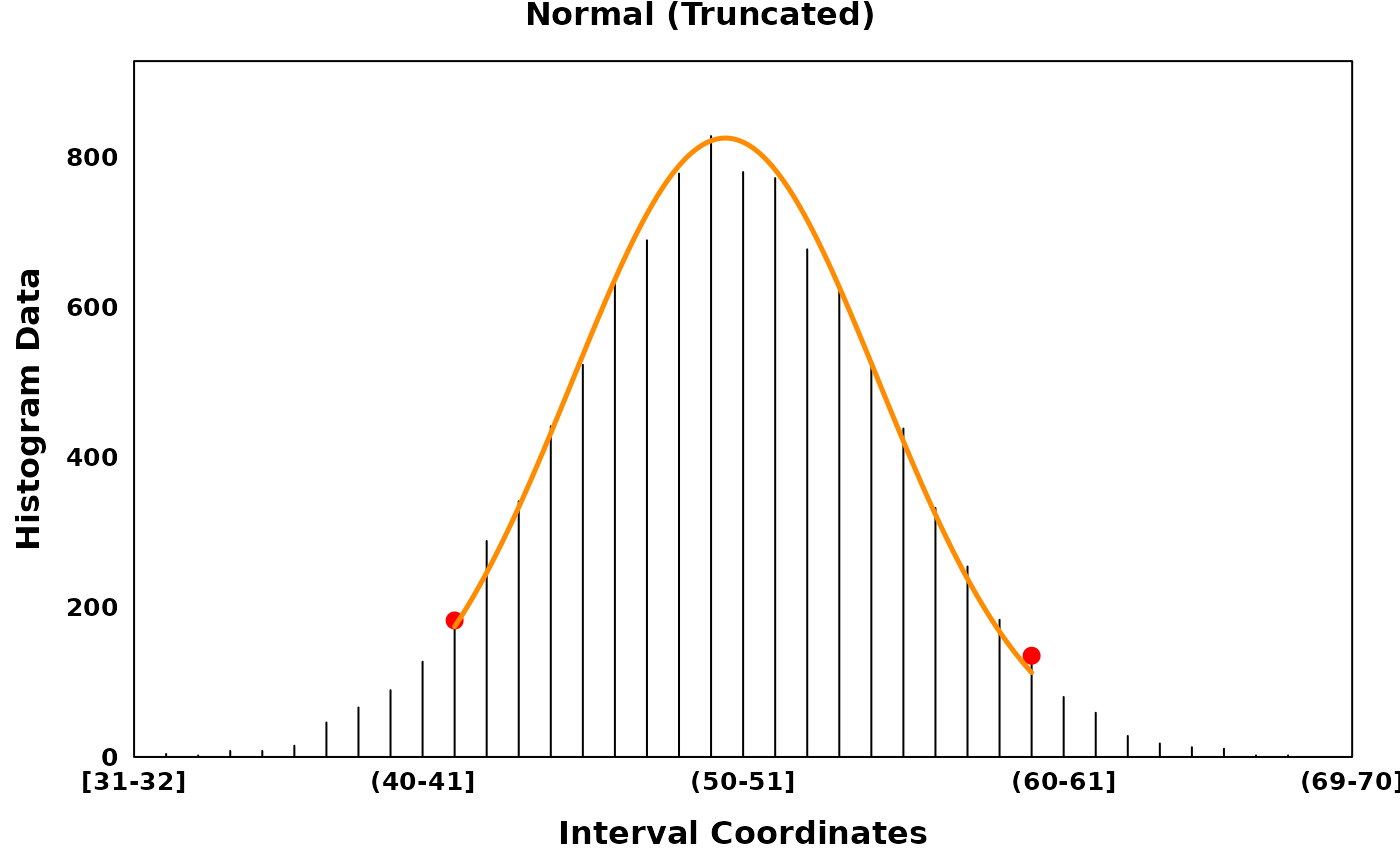
Three distributions are provided: the normal (Gaussian) distribution, the uniform distribution and the gamma distribution. The gamma distribution can be flipped to account for skew in the negative direction. Normal and gamma distributions can be fit in their full form or in a truncated form.
set.seed(314)
gaussian_data <- rnorm(10000, mean = 50, sd = 5)
gaussian_histogram <- observations_to_histogram(gaussian_data)
# Keeping all data and fitting a full model
gaussian_full <- segment_and_fit(
histogram_obj = gaussian_histogram,
eps = 1,
seed = 314,
remove_low_entropy = FALSE,
truncated_models = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
# Removing noisy data and fitting a truncated model
gaussian_truncated <- segment_and_fit(
histogram_obj = gaussian_histogram,
eps = 1,
seed = 314,
remove_low_entropy = TRUE,
truncated_models = TRUE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
gaussian_full_plt <- create_coverageplot(
gaussian_full,
main = "Normal (Full)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
gaussian_truncated_plt <- create_coverageplot(
gaussian_truncated,
main = "Normal (Truncated)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(gaussian_full_plt)
print(gaussian_truncated_plt)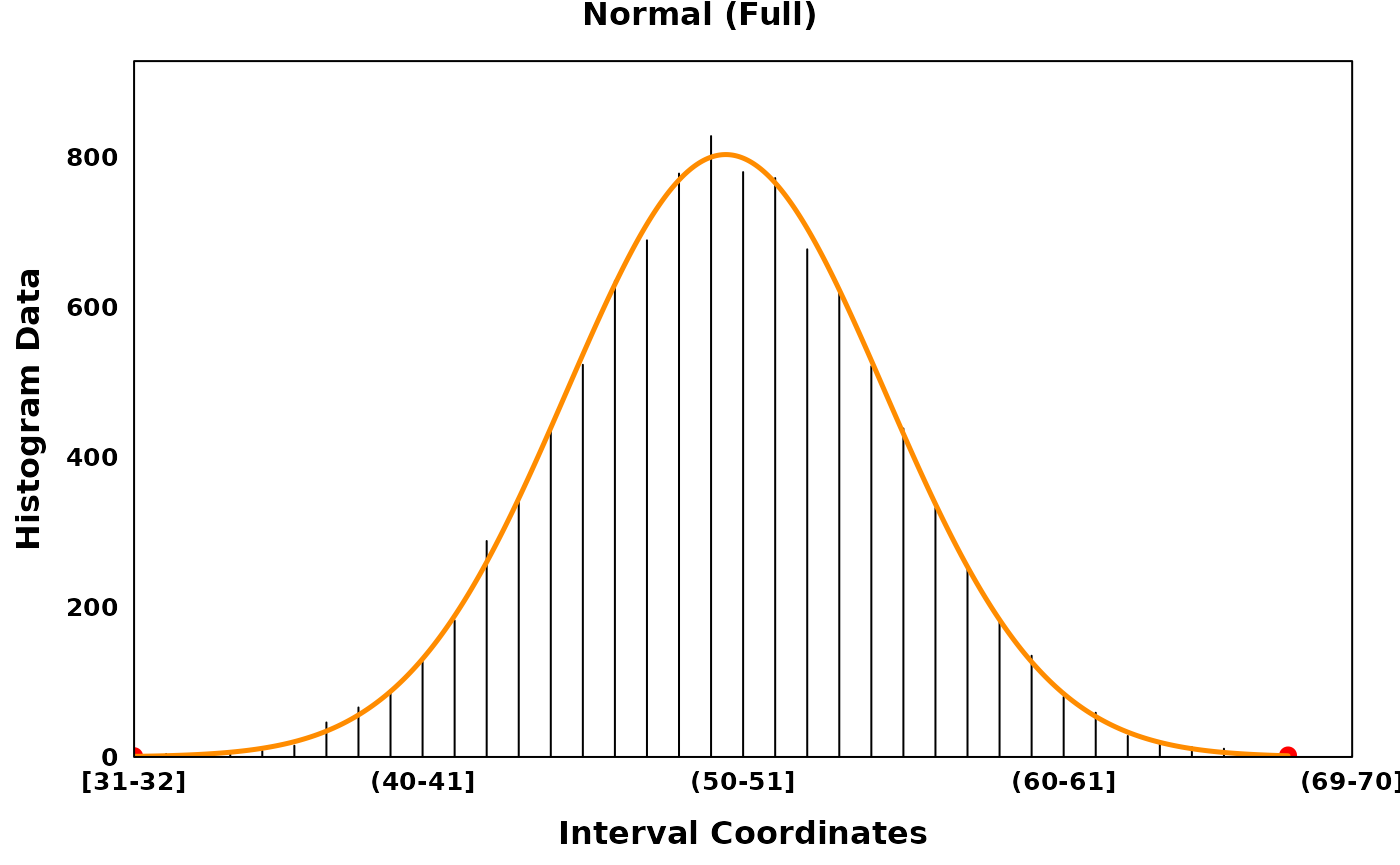
set.seed(314)
gamma_data <- rgamma(10000, shape = 2, rate = 0.4)
gamma_histogram <- observations_to_histogram(gamma_data)
# Keeping all data and fitting a full model
gamma_full <- segment_and_fit(
histogram_obj = gamma_histogram,
eps = 1,
seed = 314,
remove_low_entropy = FALSE,
truncated_models = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
# Removing noisy data and fitting a truncated model
gamma_truncated <- segment_and_fit(
histogram_obj = gamma_histogram,
eps = 1,
seed = 314,
remove_low_entropy = TRUE,
truncated_models = TRUE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
gamma_full_plt <- create_coverageplot(
gamma_full,
main = "Gamma (Full)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
gamma_truncated_plt <- create_coverageplot(
gamma_truncated,
main = "Gamma (Truncated)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(gamma_full_plt)
print(gamma_truncated_plt)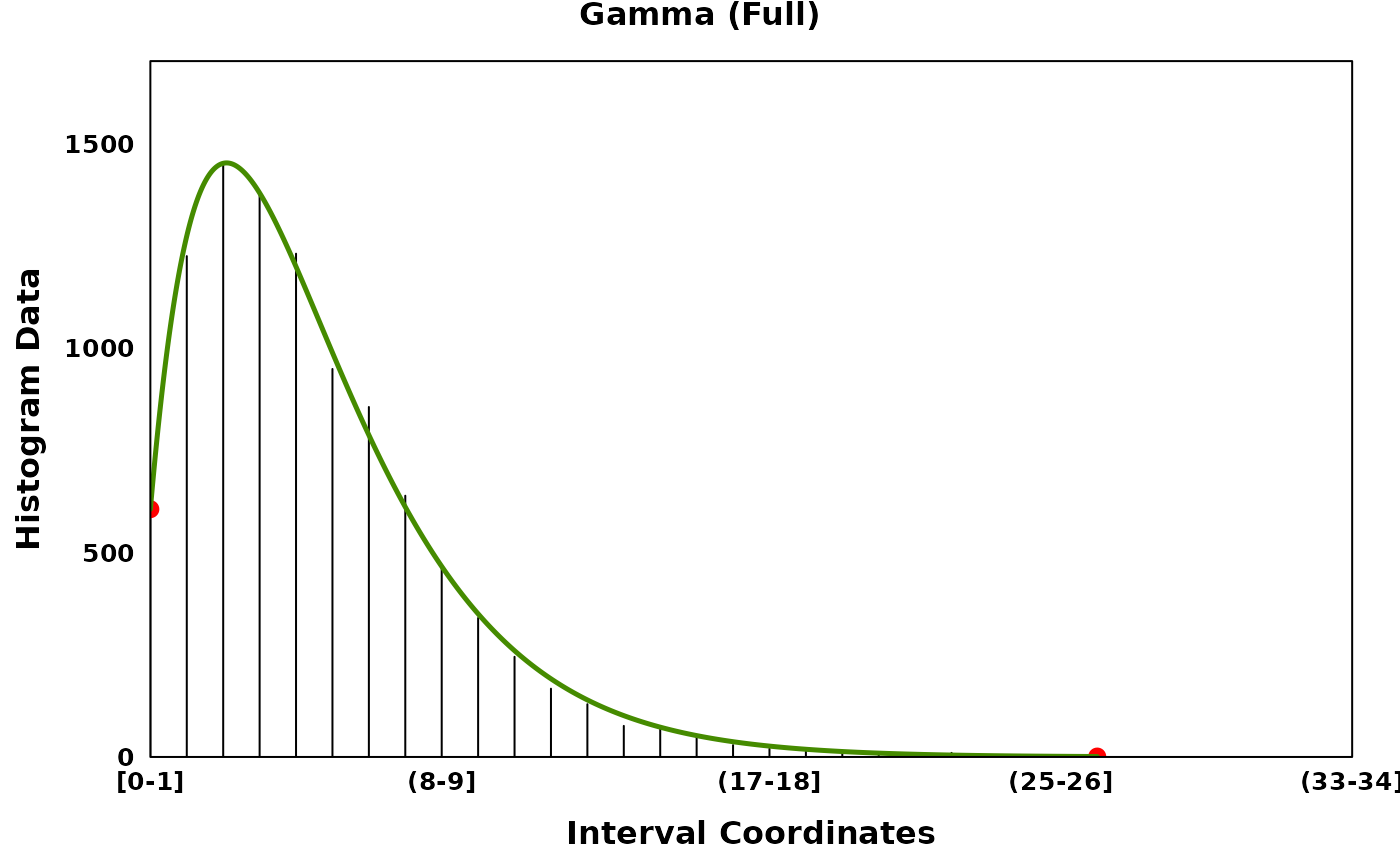
set.seed(314)
gamma_flip_data <- rgamma_flip(9000, shape = 4, rate = 0.4) + 80
gamma_flip_histogram <- observations_to_histogram(gamma_flip_data)
# Keeping all data and fitting a full model
gamma_flip_full <- segment_and_fit(
histogram_obj = gamma_flip_histogram,
eps = 1,
seed = 314,
remove_low_entropy = FALSE,
truncated_models = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
# Removing noisy data and fitting a truncated model
gamma_flip_truncated <- segment_and_fit(
histogram_obj = gamma_flip_histogram,
eps = 1,
seed = 314,
remove_low_entropy = TRUE,
truncated_models = TRUE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
gamma_flip_full_plt <- create_coverageplot(
gamma_flip_full,
main = "Gamma Flipped (Full)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
gamma_flip_truncated_plt <- create_coverageplot(
gamma_flip_truncated,
main = "Gamma Flipped (Truncated)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(gamma_flip_full_plt)
print(gamma_flip_truncated_plt)
set.seed(314)
uniform_data <- runif(10000, min = 0, max = 10)
uniform_histogram <- observations_to_histogram(uniform_data)
# Fitting a uniform segment
uniform_fit <- segment_and_fit(
histogram_obj = uniform_histogram,
eps = 1,
seed = 314,
max_uniform = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
uniform_plt <- create_coverageplot(
uniform_fit,
main = "Uniform",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(uniform_plt)
Metrics
HistogramZoo provides alternative estimators based on histogram distance for distribution parameters. Five potential metrics are available and parameters are selected to minimize the error between the density function of a statistical distribution and the observed histogram density. A full collection of histogram comparison metrics is described by (Meshgi and Ishii 2015).
Let \(h\) be our observed histogram, where \(h(i)\) is the density at bin \(i\) and \(x(i)\) be the mid-point for the bin \(i\). We define the histogram metrics, compared to a distribution \(f\) as follows:
- Jaccard index is the ratio between the intersection of the fitted and observed distributions and the union of the two distributions. When comparing two histograms, intersection is the minimum and union is the maximum.
\[ D_{J}(h, f) = 1 - \frac{\sum_{i=1}^{n}\min\left(h(i), f(x(i))\right)}{\sum_{i=1}^{n}\max\left(h(i), f(x(i))\right)} \]
- Intersection is the intersection between the fitted and observed distributions
\[ D_{\cap} = 1 - \frac{\sum_{i} \left(\min(h(i),f(x(i)) \right)}{\min\left(\vert h(i)\vert,\vert f(x(i)) \vert \right)} \]
- Kolmogorov-Smirnov statistic is the maximum divergence between the fitted and observed distributions.
\[ D_{KS}(h, f) = \max_i \left|h(i) - f(x(i))\right| \]
- Mean squared error is the mean squared difference between the fitted and observed distributions.
\[ D_{MSE}(h, f) = \frac{1}{n}\sum_{i=1}^{n}\left(h(i) - f(x(i))\right)^2 \]
- \(\chi\)-squared statistic is the \(\chi\)-squared distance between the two distributions i.e.
\[ D_{\chi^2}(h, f) = \sum_{i=1}^{n}\frac{\left[ h(i) - f(x(i)) \right]^2}{h(i) + f(x(i))} \]
Maximum likelihood estimation
Maximum likelihood estimation using histogram data (ref) is provided for distribution fitting and parameter estimation. The log likelihood of a bin is given by the following formula:
\[ \log \prod_{i = 1}^{k} (F_\theta(x_i) - F_\theta(x_0))= k\log(F_\theta(x_1) - F_\theta(x_0)) \]
where \(F_\theta\) is the cumulative distribution function of the fitted distribution with parameters \(\theta\) and \(x_i\) and \(x_0\) are the start and endpoints of a bin \((x_0, x_1]\).
Finding a consensus model
The function find_consensus_model is used to determine
the best fitting model for each segment and provides two methods of
doing so: weighted_majority_vote (weighted majority voting)
and rra (robust rank aggregation, Kolde et al. (2012)). Weighted majority voting
requires the users to provide a ranking of metrics in descending order
of importance and their associated weights and used the weighted sum of
ranks to determine the best fitting distribution. In the event of a tie,
the highest ranked metric is used to break the tie.
Maximizing uniform segments
The function identify_uniform_segment optimizes the
identification of uniform segments by iteratively trimming the ends of
the region and refitting a uniform distribution to the shortened region.
This is controlled by the threshold parameter which is the
proportion of the original region which must be preserved and the
stepsize which indicates the stepsize taken for each
search. One of the metrics must be selected to determine the best fit
while the parameter max_sd_size indicates the maximum
tolerable standard deviation of the goodness-of-fit from the best
fitting uniform distribution where the function can select the longest
uniform distribution.
set.seed(314)
# Simulating uniform data that can benefit from trimming
uniform_data <- c(
runif(10000, min = 0, max = 50),
runif(10000, min = 2, max = 48),
runif(10000, min = 4, max = 46),
runif(10000, min = 6, max = 44),
runif(10000, min = 8, max = 42)
)
uniform_histogram <- observations_to_histogram(uniform_data)
# Fitting a uniform segment
uniform_untrimmed <- segment_and_fit(
histogram_obj = uniform_histogram,
eps = 1,
seed = 314,
max_uniform = FALSE,
remove_low_entropy = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
uniform_trimmed <- segment_and_fit(
histogram_obj = uniform_histogram,
eps = 1,
seed = 314,
max_uniform = TRUE,
uniform_threshold = 0.75,
uniform_stepsize = 5,
uniform_max_sd = 1,
remove_low_entropy = FALSE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
uniform_untrimmed_plt <- create_coverageplot(
uniform_untrimmed,
main = "Uniform (Untrimmed)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
uniform_trimmed_plt <- create_coverageplot(
uniform_trimmed,
main = "Uniform (Max Uniform)",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
legend = NULL
)
print(uniform_untrimmed_plt)
print(uniform_trimmed_plt)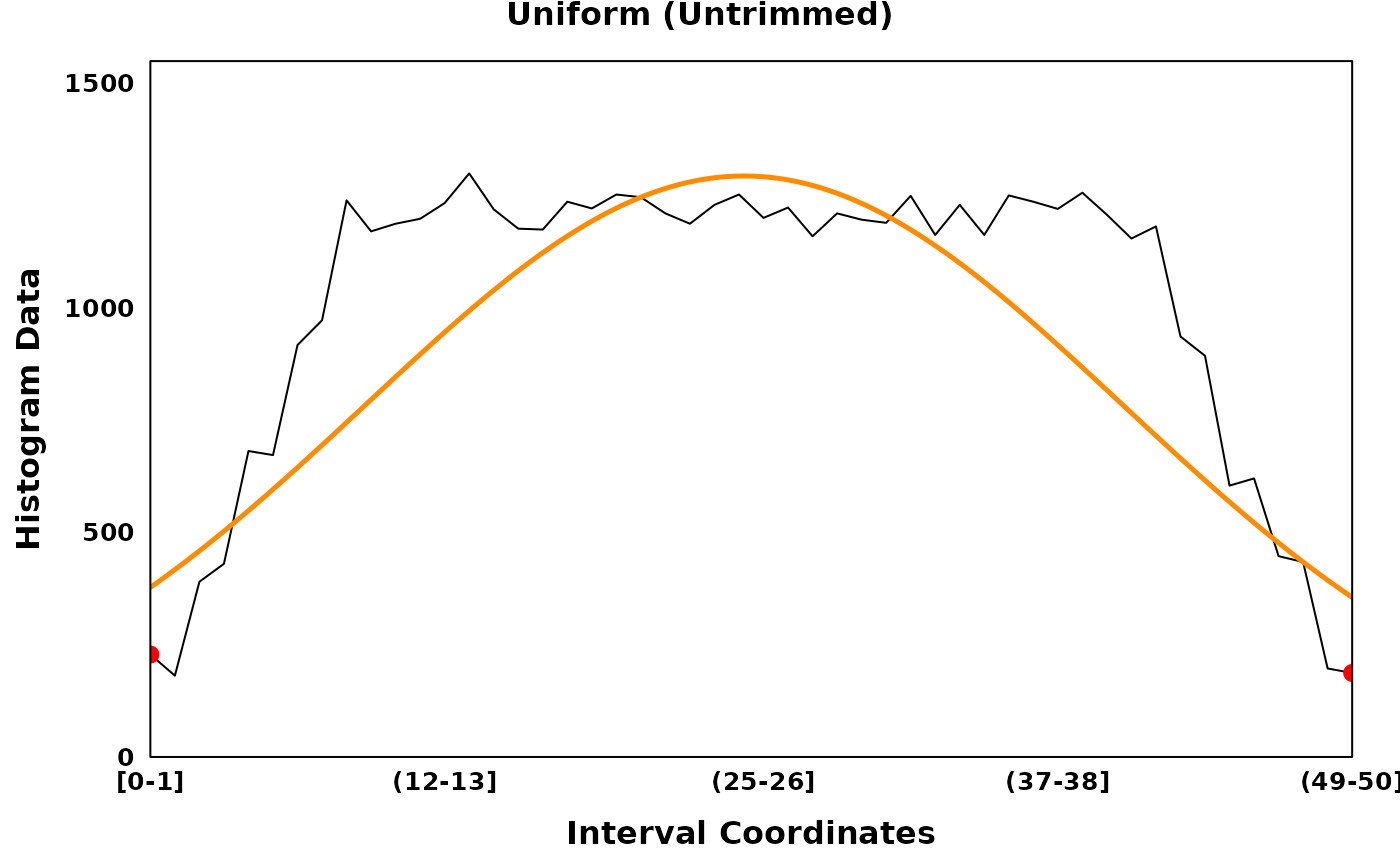

Exporting results
Summary table of results
summarize_results can be applied to a
HistogramFit object to extract the indices and summary
statistics of the analysis.
A data frame will be returned with the following columns.
- region_id character string denoting the region_id of the Histogram
- segment_id an integer id identifying the ordinal segment of the Histogram segmentation
- chr an optional column denoting the chromosome of a GenomicHistogram object
- start the interval start of the segment
- end the interval end of the segment
- strand an optional column denoting the strand of a GenomicHistogram object
- interval_count the number of intervals in the segment - used for collapsing disjoint intervals
- interval_sizes the width of each interval
- interval_starts the start index of each interval
- histogram_start The start index of the segment in the Histogram representation
- histogram_end The end index of the segment in the Histogram representation
- value the fitted value of the metric function
- metric the metric used to fit the distribution
- dist the distribution name
- sample_mean the mean of the observed histogram data
- sample_var the variance of the observed histogram data
- sample_sd the standard deviation of the observed histogram data
- sample_skew the skew of the observed histogram data
- dist_param[0-9] the values of distribution parameters
- dist_param_name[0-9] the matching names of distribution parameters
Plotting
Several plotting functions are available to aid with the
visualization of results. Plotting functions can be combined with
plotting capabilities from BoutrosLab.plotting.general
(P’ng et al. 2019) based on the
lattice R package (Sarkar, Sarkar,
and KernSmooth 2015).
create_histogram and create_coverageplot can be used to visualize the
HistogramorGenomicHistogramobjects. Currently,create_coverageplotcan be used to visualizeHistogramFitobjects, resulting fromsegment_and_fitanalysis, with annotated segmentation points and distributions.create_histogramgenerates the traditional representation of histograms as consecutive blocks whilecreate_coverageplotrepresents the histograms as lollipop plots or as lines.create_coverageplotwas designed with large histograms, such as those derived from genomic datasets, in mind.create_residualplot can be used to visualize the residuals between the fitted and observed distributions of a
HistogramFitobjectcreate_trackplot can be used to add complementary tracks to a histogram visualization when combined with
create_layerplotcreate_layerplot can be used to create a multi-panel plot with combinations of histogram, coverage, residual and track plots.
Example
A histogram is simulated from 3 statistical distributions. Two
equivalent visualizations are shown below using
create_coverageplot and create_histogram.
set.seed(314)
my_data <- c(
rnorm(8000, mean = 50, sd = 2),
rgamma(10000, shape = 2, rate = 0.4),
rgamma_flip(9000, shape = 2, rate = 0.4) + 80
)
my_histogram <- observations_to_histogram(my_data)
# A basic coverageplot to visualize the data
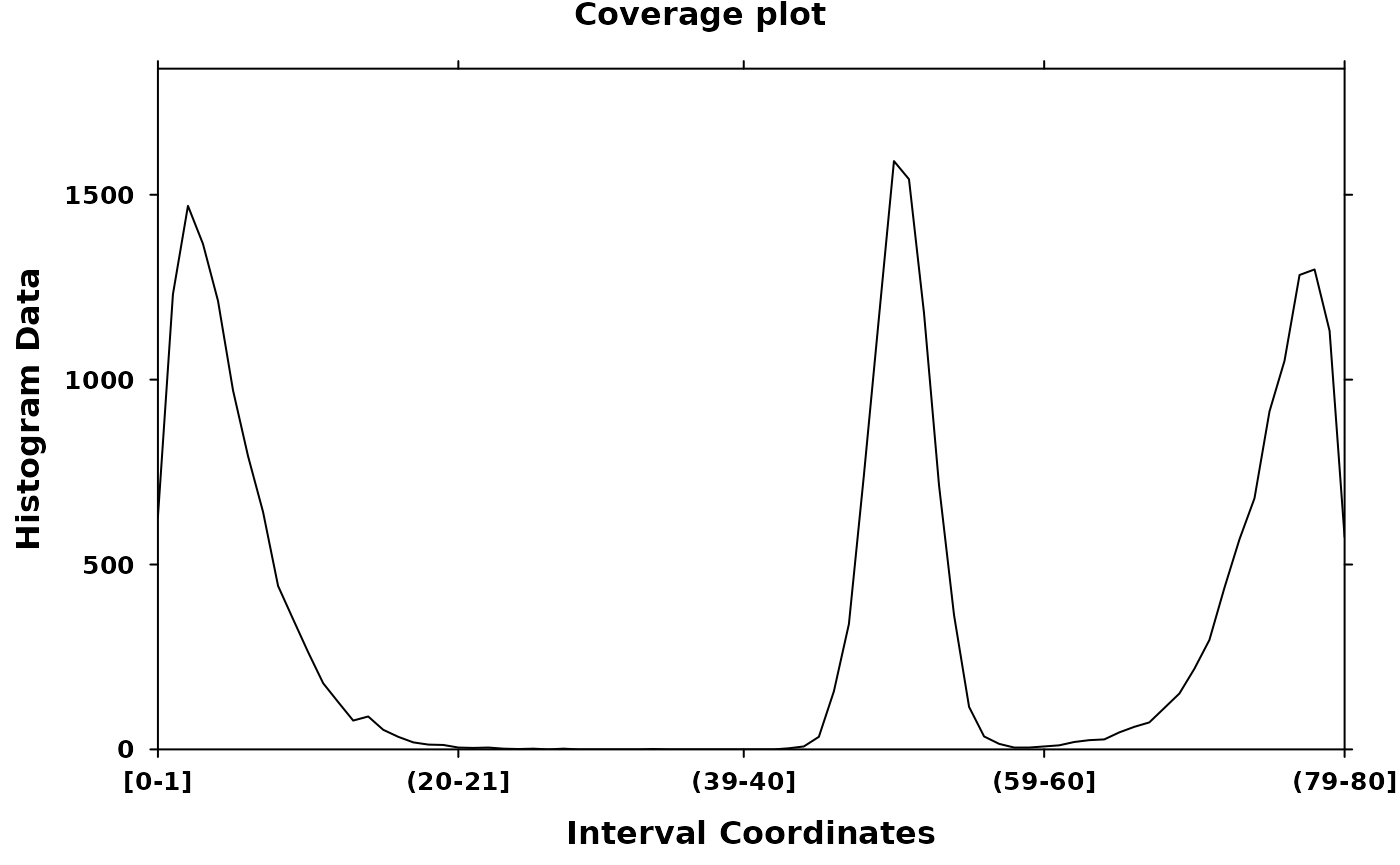
create_coverageplot(
my_histogram,
main = "Coverage plot",
main.cex = 1,
xaxis.tck = 0.5,
yaxis.tck = 0.5,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8
)
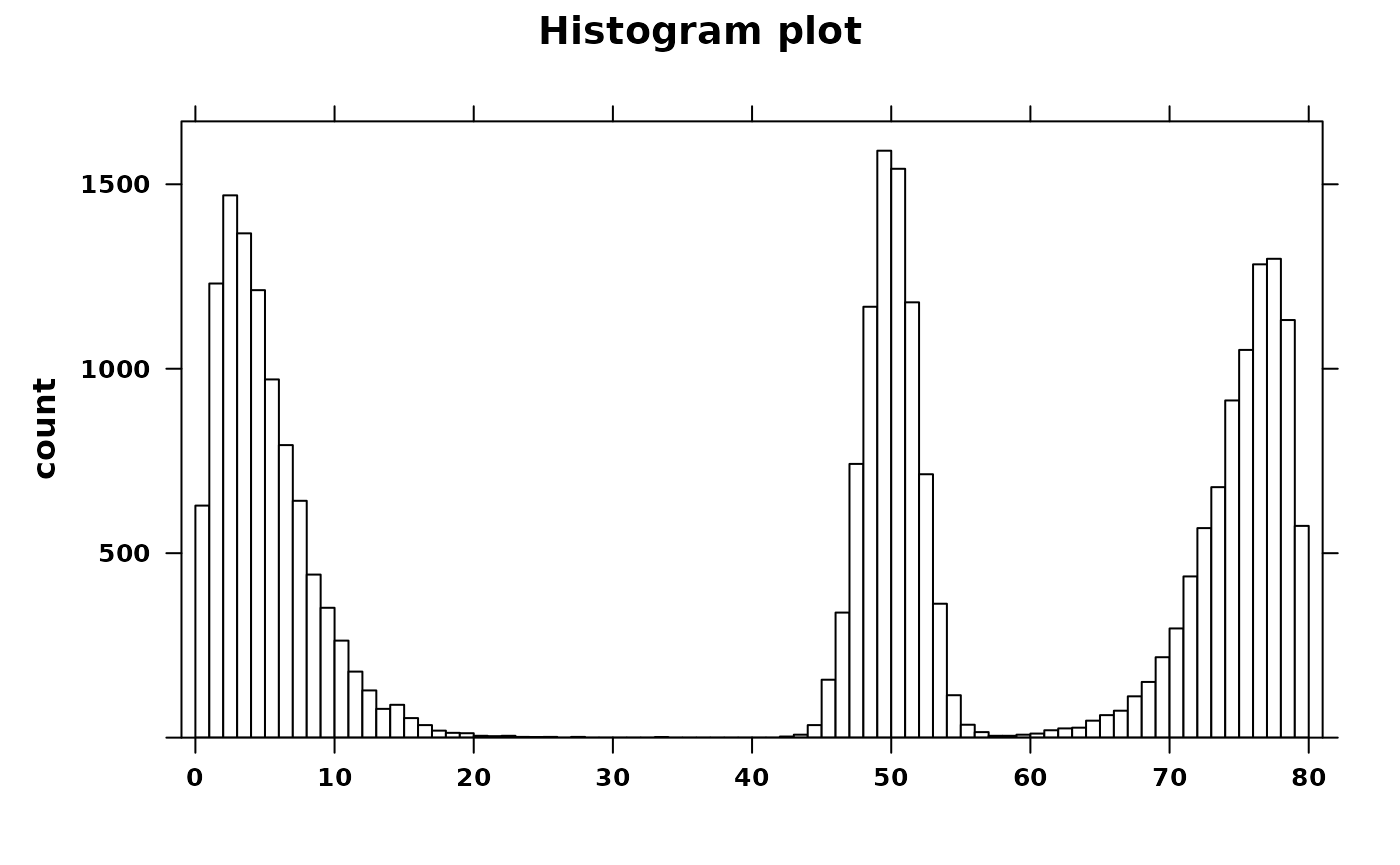
create_histogram(
my_histogram,
type = "count",
main = "Histogram plot",
main.cex = 1,
xaxis.tck = 0.5,
yaxis.tck = 0.5,
xlab.cex = 1,
ylab.cex = 1,
xaxis.cex = 0.8,
yaxis.cex = 0.8,
xat = seq(0, 80, 10)
)
HistogramZoo is applied to segment the histogram and fit distributions. Results are reported in a table.
# Conducting analysis
histogram_fit <- segment_and_fit(
histogram_obj = my_histogram,
eps = 1,
seed = 314,
truncated_models = TRUE,
metric = c("jaccard", "intersection", "ks", "mse", "chisq")
)
# summarize_results to generate
res <- summarize_results(
histogram_fit
)The summarize_results table returns a data frame with
columns describing the region_id of the original histogram
and a separate for each identified segment (segment_id).
The start and end columns indicate the segment
indices relative to the interval_start and
interval_end indices of the original histogram. The
interval_count, interval_sizes and
interval_starts columns aid in describing histograms with
disjoint intervals following a BED file
format.
knitr::kable(res[,c("region_id", "segment_id", "start", "end", "interval_count", "interval_sizes", "interval_starts")])| region_id | segment_id | start | end | interval_count | interval_sizes | interval_starts |
|---|---|---|---|---|---|---|
| 0-80 | 1 | 0 | 10 | 1 | 11, | 1, |
| 0-80 | 2 | 46 | 54 | 1 | 9, | 1, |
| 0-80 | 3 | 71 | 80 | 1 | 10, | 1, |
The histogram_start and histogram_end
columns indicate the start and end indices relative to the histogram
itself (i.e. assuming that each bin is ordinal). The value
refers to the GOF of the selected metric and the
dist column indicates the distribution fitted.
| histogram_start | histogram_end | value | metric | dist |
|---|---|---|---|---|
| 1 | 10 | 0.9807606 | consensus | gamma |
| 47 | 54 | 0.9656822 | consensus | norm |
| 72 | 80 | 0.9789559 | consensus | gamma_flip |
The sample_mean, sample_var,
sample_sd and sample_skew columns show summary
statistics estimated on the observed data for each segment.
| sample_mean | sample_var | sample_sd | sample_skew |
|---|---|---|---|
| 4.225027 | 5.872675 | 2.423360 | 0.4214018 |
| 50.000982 | 3.111514 | 1.763948 | 0.0132582 |
| 75.984753 | 5.005186 | 2.237227 | -0.3302141 |
The dist_param columns hold the values of the
distribution parameters
| dist_param1 | dist_param2 | dist_param3 | dist_param4 |
|---|---|---|---|
| 2.017559 | 0.3974044 | 0 | 0 |
| 50.007707 | 1.9119002 | 46 | 54 |
| 1.988789 | 0.3840282 | 80 | 0 |
while the dist_param_name columns hold the names of the
parameters corresponding to each distribution
knitr::kable(res[,c("dist_param1_name", "dist_param2_name", "dist_param3_name", "dist_param4_name")])| dist_param1_name | dist_param2_name | dist_param3_name | dist_param4_name |
|---|---|---|---|
| shape | rate | shift | a |
| mean | sd | a | b |
| shape | rate | offset | a |
A plot of the result is generated.
# Histogram Fit
coverage_plt <- create_coverageplot(
histogram_fit,
main = "HistogramFit",
main.cex = 1,
xaxis.tck = 0,
yaxis.tck = 0.5,
xlab.label = '',
ylab.cex = 1,
xaxis.label = NULL,
yaxis.cex = 0.8,
legend = NULL
)
residual_plt <- create_residualplot(
histogram_fit,
main = '',
xaxis.tck = 0,
yaxis.tck = 0.5,
xlab.label = '',
ylab.cex = 1,
ylab.label = "Residuals",
xaxis.label = NULL,
yaxis.cex = 0.8
)
track_plt <- create_trackplot(
res,
row_id = "region_id",
metric_id = "value",
start_id = "histogram_start",
end_id = "histogram_end",
xat = generate_xlabels(histogram_fit, return_xat = T),
xaxis.lab = generate_xlabels(histogram_fit),
main = '',
xaxis.tck = 0,
yaxis.tck = 0,
xlab.cex = 1,
ylab.cex = 1,
ylab.label = "GOF",
xaxis.cex = 0.8,
yaxis.lab = NULL
)
covariate.legend <- list(
legend = list(
colours = c(
"orange",
"chartreuse4",
"chartreuse3"
),
labels = c(
"Normal",
"Gamma",
"Gamma Flipped"
),
title = expression(bold(underline('Fitted Distributions'))),
lwd = 0.5
),
legend = list(
colours = c("red"),
labels = c("FTC Segmentation"),
title = expression(bold(underline('Points'))),
lwd = 0.5
),
legend = list(
colours = c('blue', 'red'),
labels = c(
formatC(min(res[,"value"]), digits = 2),
formatC(max(res[,"value"]), digits = 2)
),
title = expression(bold(underline('Fit'))),
continuous = TRUE,
width = 3,
tck = 1,
tck.number = 3,
at = c(0,100),
angle = -90,
just = c("center","bottom")
)
)
side.legend <- legend.grob(
legends = covariate.legend,
label.cex = 0.8,
title.cex = 0.8,
title.just = 'left',
title.fontface = 'bold',
size = 2
)
summary_figure <- create_layerplot(
plot.objects = list(coverage_plt, residual_plt, track_plt),
plot.objects.heights = c(4,1.5,1.5),
y.spacing = -3.5,
ylab.axis.padding = -13,
legend = list(
right = list(
x = 0.8,
y = 1,
fun = side.legend
)
),
right.legend.padding = 0.5,
right.padding = 1
)
# For exporting the plot
# filename = "SummaryFigure.pdf"
# pdf(filename, width = 7, height = 7)
# print(summary_figure)
# dev.off()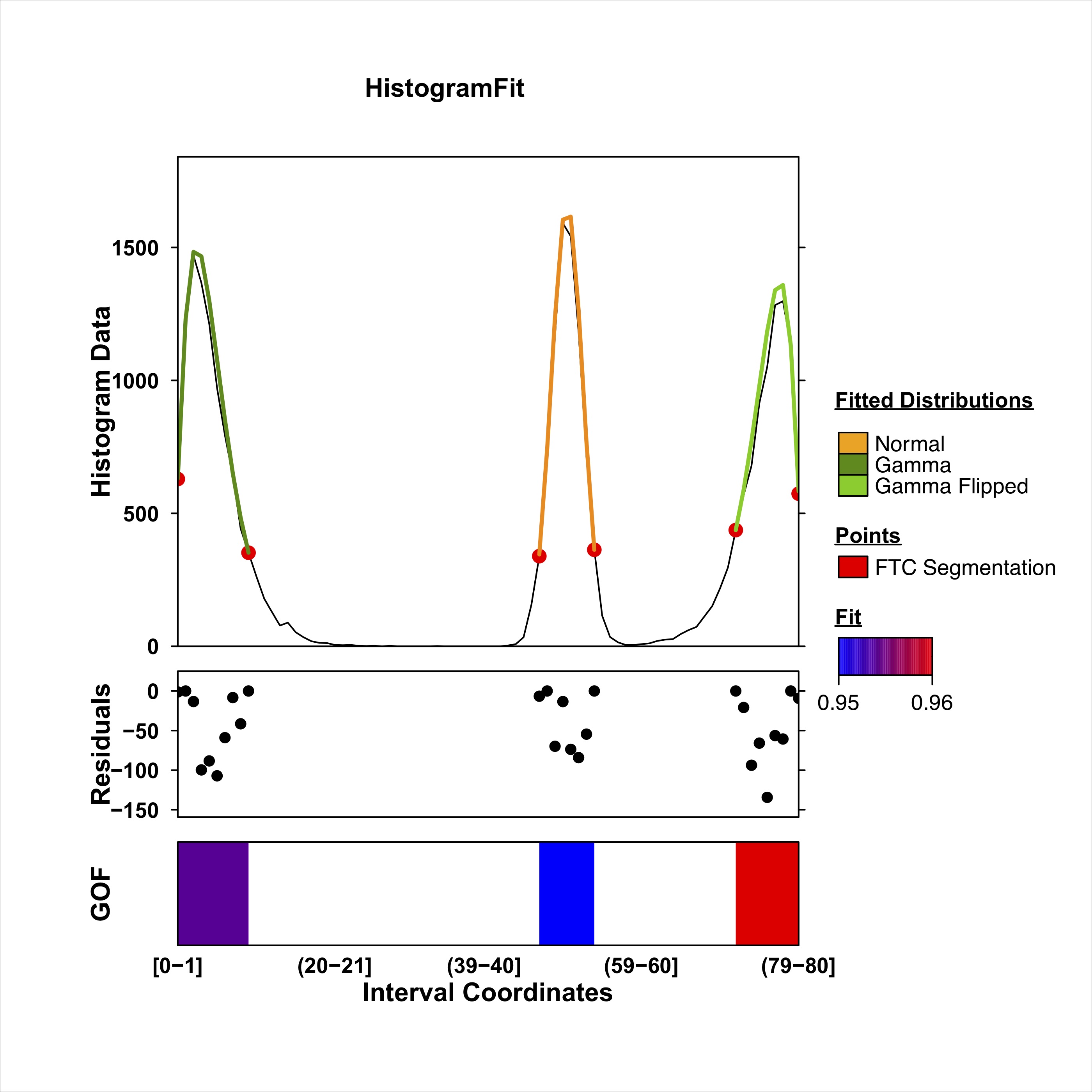
Summary Figure